


Journal publications
An efficient and accurate approach to identify similarities between biological sequences using pair amino acid composition and physicochemical properties
Lida Hooshyar, Beatriz Hernández-Jiménez, Alireza Khastan, Mahdi Vasighi
Soft Computing (2024)
Full text Abstract
Our study presents a novel method for analyzing biological sequences, utilizing Pairwise Amino Acid Composition and Amino Acid physicochemical properties to construct a feature vector. This step is pivotal, as by utilizing pairwise analysis, we consider the order of amino acids, thereby capturing subtle nuances in sequence structure. Simultaneously, by incorporating physicochemical properties, we ensure that the hidden information encoded within amino acids is not overlooked. Furthermore, by considering both the frequency and order of amino acid pairs, our method mitigates the risk of erroneously clustering different sequences as similar, a common pitfall in older methods. Our approach generates a concise 48-member vector, accommodating sequences of arbitrary lengths efficiently. This compact representation retains essential amino acid-specific features, enhancing the accuracy of sequence analysis. Unlike traditional approaches, our algorithm avoids the introduction of sparse vectors, ensuring the retention of important information. Additionally, we introduce fuzzy equivalence relationships to address uncertainty in the clustering process, enabling a more nuanced and flexible clustering approach that captures the inherent ambiguity in biological data. Despite these advancements, our algorithm is presented in a straightforward manner, ensuring accessibility to researcherswith varying levels of computational expertise. This enhancement improves the robustness and interpretability of our method, providing researchers with a comprehensive and user-friendly tool for biological sequence analysis.
Discovering SNP-disease relationships in genome-wide SNP data using an improved harmony search based on SNP locus and genetic inheritance patterns
Fariba Esmaeili, Zahra Narimani, Mahdi Vasighi
Plos one 18.10 (2023) e0292266
Full text Abstract
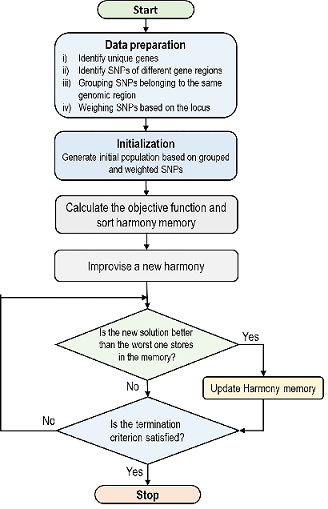
Advances in high-throughput sequencing technologies have made it possible to access millions of measurements from thousands of people. Single nucleotide polymorphisms (SNPs), the most common type of mutation in the human genome, have been shown to play a significant role in the development of complex and multifactorial diseases. However, studying the synergistic interactions between different SNPs in explaining multifactorial diseases is challenging due to the high dimensionality of the data and methodological complexities. Existing solutions often use a multi-objective approach based on metaheuristic optimization algorithms such as harmony search. However, previous studies have shown that using a multi-objective approach is not sufficient to address complex disease models with no or low marginal effect. In this research, we introduce a locus-driven harmony search (LDHS), an improved harmony search algorithm that focuses on using SNP locus information and genetic inheritance patterns to initialize harmony memories. The proposed method integrates biological knowledge to improve harmony memory initialization by adding SNP combinations that are likely candidates for interaction and disease causation. Using a SNP grouping process, LDHS generates harmonies that include SNPs with a higher potential for interaction, resulting in greater power in detecting disease-causing SNP combinations. The performance of the proposed algorithm was evaluated on 200 synthesized datasets for disease models with and without marginal effect. The results show significant improvement in the power of the algorithm to find disease-related SNP sets while decreasing computational cost compared to state-of-the-art algorithms. The proposed algorithm also demonstrated notable performance on real breast cancer data, showing that integrating prior knowledge can significantly improve the process of detecting disease-related SNPs in both real and synthesized data.
Sequence-Based Prediction of Plant Allergenic Proteins: Machine Learning Classification Approach
Miroslava Nedyalkova, Mahdi Vasighi, Amirreza Azmoon, Ludmila Naneva, and Vasil Simeonov
ACS Omega, 8 (2023) 3698
Full text Abstract

This Article proposes a novel chemometric approach to understanding and exploring the allergenic nature of food proteins. Using machine learning methods (supervised and unsupervised), this work aims to predict the allergenicity of plant proteins. The strategy is based on scoring descriptors and testing their classification performance. Partitioning was based on support vector machines (SVM), and a k-nearest neighbor (KNN) classifier was applied. A fivefold cross-validation approach was used to validate the KNN classifier in the variable selection step as well as the final classifier. To overcome the problem of food allergies, a robust and efficient method for protein classification is needed.
A multilevel approach for screening natural compounds as an antiviral agent for COVID-19
Mahdi Vasighi, Julia Romanova, Miroslava Nedyalkova
Computational Biology and Chemistry, 98 (2022) 107694
Full text Abstract
The COVID-19 has a worldwide spread, which has prompted concerted efforts to find successful drug treatments. Drug design focused on finding antiviral therapeutic agents from plant-derived compounds which may disrupt the attachment of SARS-CoV-2 to host cells is with a pivotal need and role in the last year. Herein, we provide an approach based on drug design methods combined with machine learning approaches to classify and discover inhibitors for COVID-19 from natural products. The spike receptor-binding domain (RBD) was docked with database of 125 ligands. The docking protocol based on several steps was performed within Autodock Vina to identify the high-affinity binding mode and to reveal more insights into interaction between the phytochemicals and the RBD domain. A protein-ligand interaction analyzer has been developed. The drug-likeness properties of explored inhibitors are analyzed in the frame of exploratory data analyses. The developed computational protocol yielded a comprehensive pipeline for predicting the inhibitors to prevent the entry RBD region.
Inhibition Ability of Natural Compounds on Receptor-Binding Domain of SARS-CoV2: An In Silico Approach
Miroslava Nedyalkova, Mahdi Vasighi, Subrahmanyam Sappati, Anmol Kumar, Sergio Madurga, Vasil Simeonov
Pharmaceuticals, 14 (2021) 1328
Full text Abstract
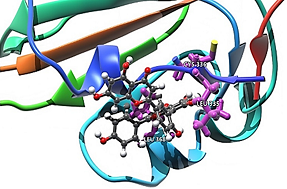
The lack of medication to treat COVID-19 is still an obstacle that needs to be addressed by all possible scientific approaches. It is essential to design newer drugs with varied approaches. A receptor-binding domain (RBD) is a key part of SARS-CoV-2 virus, located on its surface, that allows it to dock to ACE2 receptors present on human cells, which is followed by admission of virus into cells, and thus infection is triggered. Specific receptor-binding domains on the spike protein play a pivotal role in binding to the receptor. In this regard, the in silico method plays an important role, as it is more rapid and cost effective than the trial and error methods using experimental studies. A combination of virtual screening, molecular docking, molecular simulations and machine learning techniques are applied on a library of natural compounds to identify ligands that show significant binding affinity at the hydrophobic pocket of the RBD. A list of ligands with high binding affinity was obtained using molecular docking and molecular dynamics (MD) simulations for protein–ligand complexes. Machine learning (ML) classification schemes have been applied to obtain features of ligands and important descriptors, which help in identification of better binding ligands. A plethora of descriptors were used for training the self-organizing map algorithm. The model brings out descriptors important for protein–ligand interactions.
Persistent organic pollutants (POPs) - QSPR classification models by means of Machine learning strategies
Ekaterina Vakarelska, Miroslava Nedyalkova, Mahdi Vasighi, Vasil Simeonov
Chemosphere, 287 (2022) 132189
Full text Abstract
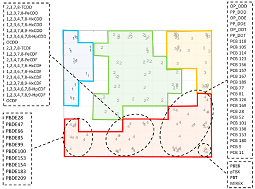
Persistent Organic pollutants (POPs) are toxic chemicals with a shallow degradation rate and global negative impact. Their physicochemical is combined with the complex effects of long-term POPs accumulation in the environment and transport function through the food chain. That is why POPs have been linked to adverse effects on human health and animals. They circulate globally via different environmental pathways, and could be detected in regions far from their source of origin. The primary goal of the present study is to carry out classification of various representatives of POPs using different theoretical descriptors (molecular, structural) to develop quantitative structure−properties relationship (QSPR) models for predicting important properties POPs. Multivariate statistical methods such as hierarchical cluster analysis, principal components analysis and self-organizing maps were applied to reach excellent partitioning of 149 representatives of POPs into 4 classes using ten most appropriate descriptors (out of 63) defined by variable reduction procedure. The predictive capabilities of the defined classes could be applied as a pattern recognition for new and unidentified POPs, based only on structural properties that similar molecules may have. The additional self-organizing maps technique made it possible to visualize the feature-space and investigate possible patterns and similarities between POPs molecules. It contributes to confirmation of the proper classification into four classes. Based on SOM results, the effect of each variable and pattern formation has been presented.
Efficient portfolio construction by means of CVaR and k-means++ clustering analysis: Evidence from the NYSE
Fazlollah Soleymani, Mahdi Vasighi
International Journal of Finance & Economics, (2020) 1-15
Full text Abstract
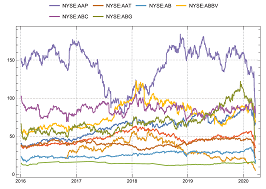
The major target of this article is to build a machine learning model furnishing an efficient and quick analysis for a large portfolio of stocks. Towards constructing such an efficient portfolio, we employ the Value-at-Risk (VaR) and Conditional Value-at-Risk (CVaR) as tools of well-consolidated use for controlling the anomalies' presence of potential danger to the financial stability of the portfolio. It is shown how the well-resulted k-means++ clustering technique is employed to cluster financial returns for the stocks of a system and then the risk measures of VaR and CVaR are obtained for the clusters to find the most and least riskiest groups of stocks. The proposed procedure is fast for clustering a financial large set of data by providing many features for each cluster.
Disease-specific protein corona sensor arrays mayhave disease detection capacity
Giulio Caracciolo, Reihaneh Safavi-Sohi, Reza Malekzadeh, Hossein Poustchi, Mahdi Vasighi, Riccardo Zenezini Chiozzi, Anna Laura Capriotti, Aldo Lagana, Mohammad Hajipour, Marina Di Domenico, Angelina Di Carlo, Damiano Caputo, Haniyeh Aghaverdi, Massimiliano Papi, Valentina Palmieri, Angela Santoni, Sara Palchetti, Luca Digiacomo, Daniela Pozzi, Kenneth S. Suslick, Morteza Mahmoudi
Nanoscale Horizons, 4 (2019) 1063-1076
Full text Abstract
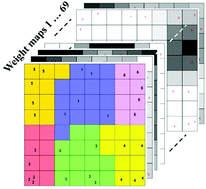
The earlier any catastrophic disease (e.g., cancer) is diagnosed, the more likely it can be treated, providing improved patient prognosis, extended survival and better quality of life. In early 2014, we revealed that various types of disease can substantially affect the composition/profile of protein corona (i.e., a layer of biomolecules that forms at the surface of nanoparticles upon their interactions with biological fluids). Here, by combining the concepts of disease-specific protein corona and sensor array technology we developed a platform with disease detection capacity using blood plasma. Our sensor array consists of three cross-reactive liposomes, with distinct lipid composition and surface charge. Rather than detecting a specific biomarker, the sensor array provides pattern recognition of the corona protein composition adsorbed on the liposomes. As a feasibility study, sensor array validation was performed using plasma samples obtained from patients diagnosed with five different cancer types (i.e. lung cancer, glioblastoma, meningioma, myeloma, and pancreatic cancer) and a control group of healthy donors. Although no single corona composition is specific for any one cancer type, overlapping but distinct patterns of the corona composition constitutes a unique "fingerprint" for each type of cancer (with a high classification accuracy, i.e. 99.4%). To finally probe the capacity of this sensor array for early detection of cancers, we used cohort plasma obtained from healthy people who were subsequently diagnosed several years after plasma collection with lung, brain, and pancreatic cancers. Our results suggest that the disease-specific protein corona sensor array will not only be instrumental in the screening, detection, and identification of diseases, but may also help identify novel protein pattern markers whose role in disease development and/or disease biology has not been appreciated so far.
Community Detection in Complex Networks by Detecting and Expanding Core Nodes Through Extended Local Similarity of Nodes
Kamal Berahmand, Asgarali Bouyer, Mahdi Vasighi
IEEE Transactions on Computational Social Systems 5, no.4 (2018): 1021-1033.
Full text Abstract
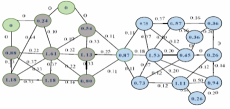
As the community detection is able to facilitate the discovery of hidden information in complex networks, it has been drawn a lot of attention recently. However, due to the growth in computational power and data storage, the scale of these complex networks has grown dramatically. In order to detect communities by utilizing global approaches, it is required to have all the global information of the whole network; something which is impossible, because of the rapid growth in the size of the networks.
In this paper, a local approach has been proposed based on the detection and expansion of core nodes. First, a community's central node (core node) which has a high level of embeddedness is detected based on the similarity between graph's nodes. By using this, the total weights of a weighted graph's edges created. Following by that, the expansion of these nodes will be considered, by utilizing the concept of node's membership based on the definition of strong community for weighted graphs. It can be seen that in detecting communities, the more accurate the weights of edges detected based on the node similarity, the more precise the local algorithm will be. In fact, the algorithm has the ability to detect all the graph's communities in a network using local information as well as identifying various roles of nodes, either being (core or outlier). Test results on both real-world and artificial networks prove that the quality of the communities which are detected by the proposed algorithm is better than the results which are achieved by other state-of-the-art algorithms in the complex networks.
A Directed Batch Growing Approach to Enhance the Topology Preservation of Self-Organizing Map
Mahdi Vasighi, Homa Amini
Applied Soft Computing 55 (2017) 424-435.
Full text Preprint Abstract
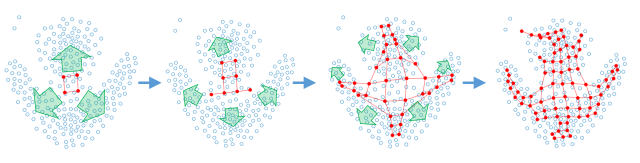
The growing self-organizing map (GSOM) possesses effective capability to generate feature maps and visualizing high-dimensional data without pre-determining their size. Most of the proposed growing SOM algorithms use an incremental learning strategy. The conventional growing approach of GSOM is based on filling all available position around the candidate neuron which can decrease the topology preservation quality of the map due to the misconfiguration and twisting of the map which could be a consequence of unexpected network growth and improper neuron addition and weight initialization. To overcome this problem, in this paper we introduce a batch learning strategy for growing self-organizing maps called DBGSOM which direct the growing process based on the accumulative error around the candidate boundary neuron. In the proposed growing approach, just one new neuron is added around each candidate boundary neuron. The DBGSOM offers suitable mechanisms to find a proper growing positions and allocating initial weight vectors for the new neurons.
The potential of the DBGSOM was investigated with one synthetic dataset and six real-world benchmark datasets in terms of topology preservation and mapping quality. Experimental results showed that the proposed growing strategy provides an enhanced topology preserved map and reduces the susceptibility of twisting compared to GSOM. Furthermore, the proposed method has a better clustering ability than GSOM and SOM. According to the lower number of neurons generated by DBGSOM, it needs less time to learn the manifold of the data points compared to GSOM.
Structural classification of proteins using texture descriptors extracted from the cellular automata image
Hamidreza Kavianpour, Mahdi Vasighi
Amino Acids 49 (2017) 261-271.
Full text Preprint Datasets Abstract
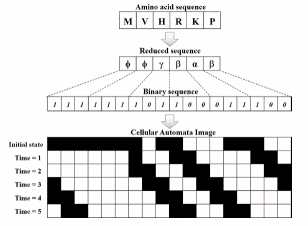
Nowadays, having knowledge about cellular attributes of proteins has an important role in pharmacy, medical science and molecular biology. These attributes are closely correlated with the function and three-dimensional structure of proteins. Knowledge of protein structural class is used by various methods for better understanding the protein functionality and folding patterns. Computational methods and intelligence systems can have an important role in performing structural classification of proteins. Most of protein sequences are saved in databanks as characters and strings and a numerical representation is essential for applying machine learning methods.
In this work, a binary representation of protein sequences is introduced based on reduced amino acids alphabets according to surrounding hydrophobicity index. Many important features which are hidden in these long binary sequences can be clearly displayed through their cellular automata images. The extracted features from these images are used to build a classification model by support vector machine. Comparing to previous studies on the several benchmark datasets, the promising classification rates obtained by tenfold cross-validation imply that the current approach can help in revealing some inherent features deeply hidden in protein sequences and improve the quality of predicting protein structural class.
Oblique rotation of factors: A novel pattern recognition strategy to classify fluorescence excitation-emission matrices of human blood plasma for early diagnosis of colorectal cancer
Mohammad Shahbazy, Mahdi Vasighi, Mohsen Kompany-Zareh, Davide Ballabio
Molecular BioSystems 12 (2016) 1963-1975.
Full text Preprint Abstract
Colorectal cancer (CRC) ranks high in both men and women, accounting for about 13% of all cancers. In this study, a novel pattern recognition strategy is proposed to improve early diagnosis of CRC through visualizing the relationship between different spectral patterns in a case-control research. Partial least squares-discriminant analysis (PLS-DA) and supervised Kohonen network (SKN) were used to classify the fluorescence excitation-emission matrices (EEMs) from 289 human blood plasma samples containing CRC patients, adenomas tumor, other non-malignant findings and healthy individuals. To obtain optimal factors, oblique rotation (OR) and genetic algorithm (GA) were used to rotate the factors by optimizing transformation matrix elements. Transformed factors were introduced to SKN to build a classification model and the model performance was examined via comparison with a common classifier; PLS-DA. Classification models were built for CRC-healthy and adenomas-healthy samples and the best results were obtained through applying GA-OR on PLS factors and introducing them to the classifiers. Nonerror rates for SKN and PLS-DA models assisted with GA (for selecting more informative PLS factors) and OR were equal to 0.97 and 0.95 in cross validation and 0.93 and 0.90 for prediction of the external test set, respectively. Moreover, according to the acceptable results for adenomas-healthy cases using optimal factors, CRC can be diagnosed in early stages. Combining classifiers and optimal factors proved to be efficient for distinguishing healthy and malignant samples, and OR can significantly improve performance of the classification model.
Diagnosis of coronary heart disease based on 1H NMR spectra of human blood plasma using genetic algorithm-based feature selection
Mahdi Vasighi, Ali Zahraei, Saeed Bagheri, Jamshid Vafaeimanesh,
Journal of Chemometrics 27 (2013) 318.
Full text Abstract
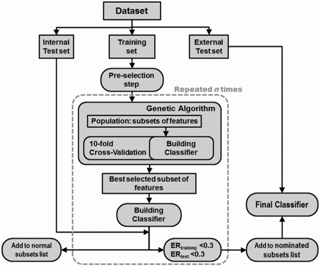
1H NMR spectroscopy was used for the diagnosis of coronary heart disease (CHD) by using human blood plasma samples. One-dimensional 1H NMR spectra from 29 normal and 35 CHD patients were obtained and investigated. Classification model was built on the basis of linear discriminant analysis in order to establish adequate model for discrimination between pathological and normal samples. Because of high similarity between 1H NMR spectra of healthy samples and patients, a feature-selection method can be used to reduce complexity of the model and improve the classification performance of the built classifier. In this paper, we presented a genetic algorithm (GA) based feature-selection method to find informative features that play a significant role in discrimination of samples. Selected subsets from multiple GA runs were used to build a classifier. The most informative features were selected according to classification performance of classifier for training and internal test set samples. The results of analysis showed that our approach can be used to improve discriminating power of classification model and simultaneously identify the important features for the diagnosis purpose and can be used in the diagnosis of CHD in patients without employing any angiographic technique.
Effects of supervised Self Organising Maps parameters on classification performance
Davide Ballabio, Mahdi Vasighi, Peter Filzmoser
Analytica Chimica Acta 765 (2013) 45.
Full text Abstract
Self Organising Maps (SOMs) are one of the most powerful learning strategies among neural networks algorithms. SOMs have several adaptable parameters and the selection of appropriate network architectures is required in order to make accurate predictions. The major disadvantage of SOMs is probably due to the network optimisation, since this procedure can be often time-expensive. Effects of network size, training epochs and learning rate on the classification performance of SOMs are known, whereas the effect of other parameters (type of SOMs, weights initialisation, training algorithm, topology and boundary conditions) are not so obvious. This study was addressed to analyse the effect of SOMs parameters on the network classification performance, as well as on their computational times, taking into consideration a significant number of real datasets, in order to achieve a comprehensive statistical comparison. Parameters were contemporaneously evaluated by means of an approach based on the design of experiments, which enabled the investigation of their interaction effects. Results highlighted the most important parameters which influence the classification performance and enabled the identification of the optimal settings, as well as the optimal architectures to reduce the computational time of SOMs.
A MATLAB toolbox for Self Organizing Maps and supervised neural network learning strategies
Davide Ballabio, Mahdi Vasighi
Chemometrics and Intelligent Laboratory Systems 118 (2012) 24.
Full text Abstract
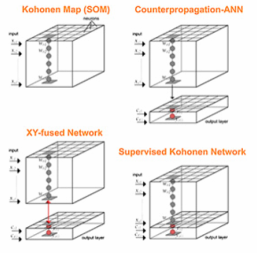
Kohonen maps and Counterpropagation Neural Networks are two of the most popular learning strategies based on Artificial Neural Networks. Kohonen Maps (or Self Organizing Maps) are basically self-organizing systems which are capable to solve the unsupervised rather than the supervised problems, while Counterpropagation Artificial Neural Networks are very similar to Kohonen maps, but an output layer is added to the Kohonen layer in order to handle supervised modelling. Recently, the modifications of Counterpropagation Artificial Neural Networks allowed introducing new supervised neural network strategies, such as Supervised Kohonen Networks and XY-fused Networks.
In this paper, the Kohonen and CP-ANN toolbox for MATLAB is described. This is a collection of modules for calculating Kohonen maps and derived methods for supervised classification, such as Counterpropagation Artificial Neural Networks, Supervised Kohonen Networks and XY-fused Networks. The toolbox comprises a graphical user interface (GUI), which allows the calculation in an easy-to-use graphical environment. It aims to be useful for both beginners and advanced users of MATLAB. The use of the toolbox is discussed here with an appropriate practical example.
Classification Ability of Self Organizing Maps in Comparison with Other Classification Methods
Mahdi Vasighi, Mohsen Kompany-Zareh
MATCH Communications in Mathematical and in Computer Chemistry 70 (2013) 29.
Full text AbstractGenetic Algorithms for architecture optimisation of Counter-Propagation Artificial Neural Networks
Davide Ballabio, Mahdi Vasighi, Viviana Consonni, Mohsen Kompany-Zareh
Chemometrics and Intelligent Laboratory Systems 105 (2011) 56.
Full text Abstract
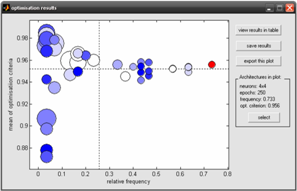
Counter-Propagation Artificial Neural Networks (CP-ANNs) require an optimisation step in order to choose the most suitable network architecture. In this paper, a new strategy for the selection of the optimal number of epochs and neurons of CP-ANNs was proposed. This strategy exploited the ability of Genetic Algorithms to optimise network parameters. Since both Genetic Algorithms and CP-ANNs can lead to overfitting, the proposed approach was developed taking into considerable account the validation of the multivariate models.
Moreover, a new criterion for calculating the Genetic Algorithm fitness function was introduced. The percentage of correctly assigned samples for calibration and internal validation were both used in the optimisation procedure, in order to get simultaneously predictive and not overfitted models. The optimisation strategy was tested by the use of several chemical benchmark data sets for classification tasks and results were compared with those of the exhaustive searching of all the possible solutions.
Nuclear magnetic resonance-based screening of thalassemia and quantification of some hematological parameters using chemometric methods
Mohammad Arjmand, Mohsen Kompany-Zareh, Mahdi Vasighi, Nastran Parvizzadeh, Zahra Zamani, Fereshteh Nazgooei
Talanta 81 (2010) 1229-1236
Full text Abstract
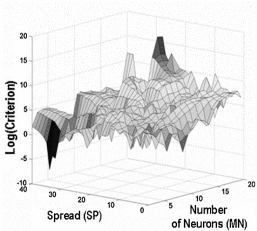
High-resolution 1H NMR spectroscopy of biofluids is a good representation of metabolic pattern and offers a high potential noninvasive technique for pathological diagnosis. Diagnosis of thalassemia and quantification of some blood parameters can be performed by using 1H NMR spectra of human blood serum in parallel with chemometric techniques. Spectra of 28 samples were collected from 15 adult male and female thalassemia patients as experimental set and 13 healthy volunteers as control set. Principal component analysis (PCA) as a dimension reduction tool was used for transforming spectra to abstract factors. The abstract factors were introduced to linear discriminant analysis (LDA), which is a common technique for classification, in order to establish adequate model for discrimination of healthy and unhealthy samples. In addition, these abstract factors were used for calibration of some blood parameters using radial basis function neural network (RBFNN) as an artificial intelligence modeling method. Different test sets (left out samples in training algorithm) were used for evaluating the quality and robustness of the built models. PCA abstract factors were employed as input for LDA model and successfully classified all the members of the test sets except one member of third test set. RBFNN also has a good capability for modeling the most of blood parameters according to proposed network parameters optimization procedure.Weconclude that 1HNMR spectroscopy, LDA and RBFNN assisted by PCA provide a powerful method for thalassemia diagnosis and prediction of some blood variants.
Analysis of solvents mixtures employing rank annihilation factor analysis on near infrared spectral data from sequential addition of analyte
Mohsen Kompany-Zareh, Mahdi Vasighi
Fuel Processing Technology 91 (2010) 62-67.
Full text Abstract
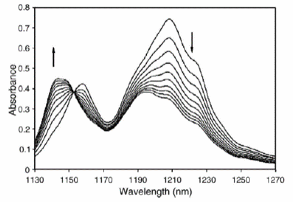
Determination of the analyte concentration in the presence of unknown interfering species is the goal in chemometrics techniques such as rank annihilation factor analysis (RAFA). In this work, using a hard model for sequential addition of analyte to an unknown mixture and a RAFA based approach, concentration of one component in a mixture of organic solvents was determined. Application of hard models causes a unique result from RAFA. The considered analyte was added sequentially to the unknown mixture in a number of steps and a near-infrared (NIR) spectrum was measured in each step. The required information for performing the analysis is the pure spectrum of the analyte (the desired solvent) and density (g mL-1) of mixtures obtained from each step of addition. Acceptable results from analysis of simulated data and binary mixture of toluene and cyclohexene, as a synthetic sample, were obtained. Effect of extent of spectral overlap, noise and initial mass fraction of analyte on accuracy and precision of the obtained results were investigated. Quantification of toluene in Gasoline, as a real sample with unknown composition, was successfully performed by the proposed method.
Resolution of near-infrared spectral data from distillation of binary mixtures and calculation of band boundaries of feasible solutions for species profiles
Mohsen Kompany-Zareh, Mahdi Vasighi
Fuel Processing Technology 89 (2008) 203-213.
Full text Abstract
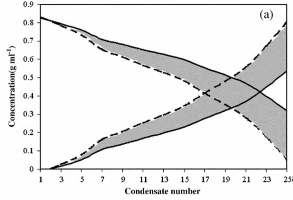
In this work, mean centering, ordinary and incomplete rank annihilation based methods were applied to estimate concentration profiles (g/mL) and pure spectra of components from an evolutionary near infrared spectral data for successive condensates from distillation process of binary mixtures. Constraints such as non-negativity, selectivity of some spectral regions and density of condensates were applied during the resolution of some series of data. Fixed size moving window evolving factor analysis (FSMWEFA) and orthogonal projection analysis (OPA) were the applied chemometrics methods for assigning the selective regions. No pure spectrum from any of components or calibration samples was necessary for performing the analysis. Three binary mixtures containing toluene:n-hexane, toluene:cyclohexene and toluene:ethanol were investigated using the proposed method. Band boundaries of feasible solutions for pure absorption spectra and species concentration profiles for the mixture of components were successfully estimated in presence of high spectral overlap. In the first case the solution was unique, but in the second and third distillations a number of acceptable solutions were obtained as band boundaries.
Conference publications
A Dynamic MOLMAP Approach for Pattern Classification in Three-Way Data
Mahdi Vasighi, Mahboubeh Talebi, Davide Ballabio
Iranian Joint Congress on Fuzzy and Intelligent Systems (CFIS) 2018, Kerman, Iran.
Full textMultiple growing self-organizing map for data classification
Mahdi Vasighi, Samira Abbasi
International Symposium on Artificial Intelligence and Signal Processing (AISP) 2017, Shiraz, Iran.
Full textPrediction of Residue Depth Level in Protein Sequence using Support Vector Machine
Mohammad Abbasshabihi, Mahdi Vasighi
The 6th Iranian Conference on Bioinformatics (2016), Tehran, Iran.
Application of self-organizing maps in biomedical image analysis using generalized cooccurrence matrix
Leila Jafari, Mahdi Vasighi, Bahram Sadeghi Bigham
The 6th Iranian Conference on Bioinformatics (2016), Tehran, Iran.
Structural Classification of Proteins using Fuzzy Cluatering and Cellular Automata
Hamidreza Kavianpour, Mahdi Vasighi
International Conference on Mathematics of Fuzziness (2016), Zanjan, Iran.
Full textGene Selection using Tabu Search in Prostate Cancer Microarray Data
Farzane Yahyanejad, Mahdi Vasighi, Angeh Aslanian, Bahareh khazaei
Contemporary Issues in Computer and Information Sciences 2012 (CICIS12), Zanjan, Iran.
Full textTOMSAGA: TOolbox for Multiple Sequence Alignment using Genetic Algorithm
Farshad B. Moghaddam, Mahdi Vasighi
Contemporary Issues in Computer and Information Sciences 2012 (CICIS12), Zanjan, Iran.
Full textComprehensive metabonomic study of malaria parasite Plasmodium falciparum by nuclear magnetic resonance spectroscopy
M. Arjmand, Z. Zamani, S. Sadeghi, M. Vasighi, S. Javadian, F. Pourfallah, F. A. Akbari Irvani, N. Parvizzadeh, R. Mohabati, F. Nazgooei,
Metabolimics 2010, Amsterdam, Netherlands.
Determination the risk of Coronary Heart Disease (CHD) using 1H NMR spectra of plasma lipoprotein with the help of pattern recognition techniques
M. Arjmand, T. Nikravesh, M. Vasighi, M.K. Rofoei, A. Movahed, N. Parvizzadrh, M. Kompany-Zareh, F. Nazgooei
Clin Chem Lab Med 2009; 47, Special Supplement, pp S1 - S409, June 2009
Metabolism-based diagnosis of beta thalasssemia using 1H NMR spectrum of human blood serum and linear vector quantization
M. Arjmand, M. Kompany-Zareh, M. Vasighi, N. Parvizzadeh, Z. Zamani, F. Nazgooei,
Clin Chem Lab Med 2009; 47, Special Supplement, pp S1 - S409, June 2009